Critical factors to the success of your clinical study
Submitting a clinical study is a process that involves several steps that must be carefully followed for its successful approval and implementation

Clinical studies in Portugal
Clinical trials of human medicines and clinical investigations of medical devices are an important step in the innovation and development process. This involves rigorous protocol design to ascertain efficacy/performance (in the case of medical devices) and safety in humans.
In the last few years, Portugal has been changing its legislation in line with European guidelines, and seeking to create initiatives and entities that aim for the agility and ethical rigor of clinical research processes. Thanks to this more favorable regulatory context and the commitment of several sectors of society, there has been a positive evolution in the conduction of clinical studies in Portugal. You read up about this evolution in more detail in Portugal in Numbers.
The execution of clinical trials in the European Union (EU) was recently subject to a profound change when the Clinical Trials Regulation (Regulation (EU) no. 536/2014) came into force on January 31st, 2022. This regulation harmonizes the submission, evaluation and supervision processes for clinical trials in the EU through the Clinical Trials Information System (CTIS) which is the single entry point for the submission of information relating to clinical trials in the EU and the European Economic Area (EEA). The CTIS functions as a public website that provides the general public with detailed information about extended clinical trials in the EU and EEA from the moment requested applications are approved in the CTIS. Furthermore, the CTIS has a working environment for clinical trial promoters or their representatives and a working environment for authorities from EU Member States and EEA countries and the European Commission.
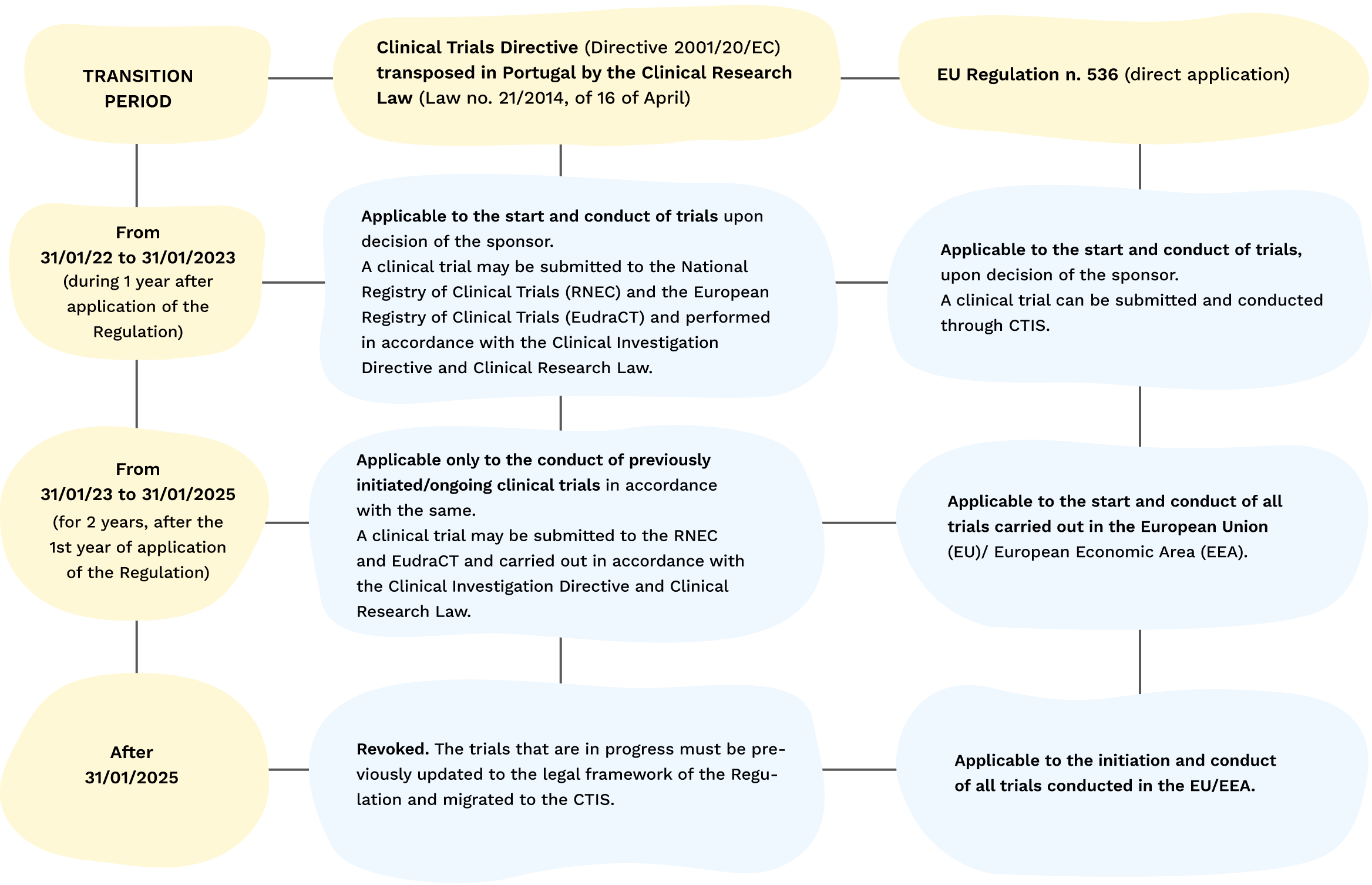
The Clinical Trials Regulation foresees a three-year transition period for the CTIS:
- From January 31st 2022 to January 30th 2023, clinical trial promoters were able to choose to submit their applications for clinical trials under the Clinical Trials Directive (Directive 2001/20/EC) on a national level, or else through the CTIS under the Clinical Trials Regulation;
- From January 31st 2023, all new clinical trial applications in the EU and EEA are submitted under the Clinical Trials Regulation via the CTIS;
- Up to January 30th 2025, trials approved under the Clinical Trials Directive that are still ongoing will have to be transferred to the procedure under the Clinical Trials Regulation and to the CTIS.
In Portugal, in addition to this Regulation, the national legislation required by Law no. 21/2014, of April 16th (Clinical Research Law), in its current wording by the Law no. 73/2015 of July 27th and Law nº 49/2018, of August 14th. Besides this national legislation, medicines for human use and medical devices are governed by additional regulations. You can refer to the most relevant regulatory information on medicines for human use and medical devices in the table below:
Regulation
Clinical trials on medicinal products for human use
-
Competent National Authority
- INFARMED, I.P.
-
Competent Ethics Committee
- CEIC
- Submission Channel
- European Legislation
- National Legislation
- Complementary Legislation
Clinical Investigations of Medical Devices
-
Competent National Authority
- INFARMED, I.P.
-
Competent Ethics Committee
- CEIC
- Submission Channel
- European Legislation
- National Legislation
- Complementary Legislation
Entities involved
For a clinical trial to be conducted in Portugal, a favorable opinion must be given by the responsible Ethics Committee in Portugal, the CEIC, and authorization must be given by the Director Council of INFARMED, I.P..
To implement a clinical study, including a clinical trial, the sponsor uses Clinical Research Centers, present in hospitals or health/research institutions with material, technical, human resources, a Clinical Ethics Committee, specialized doctors, clinical study coordinators, nurses, and pharmacists. Together, they guarantee compliance with good clinical practices and ethical principles.
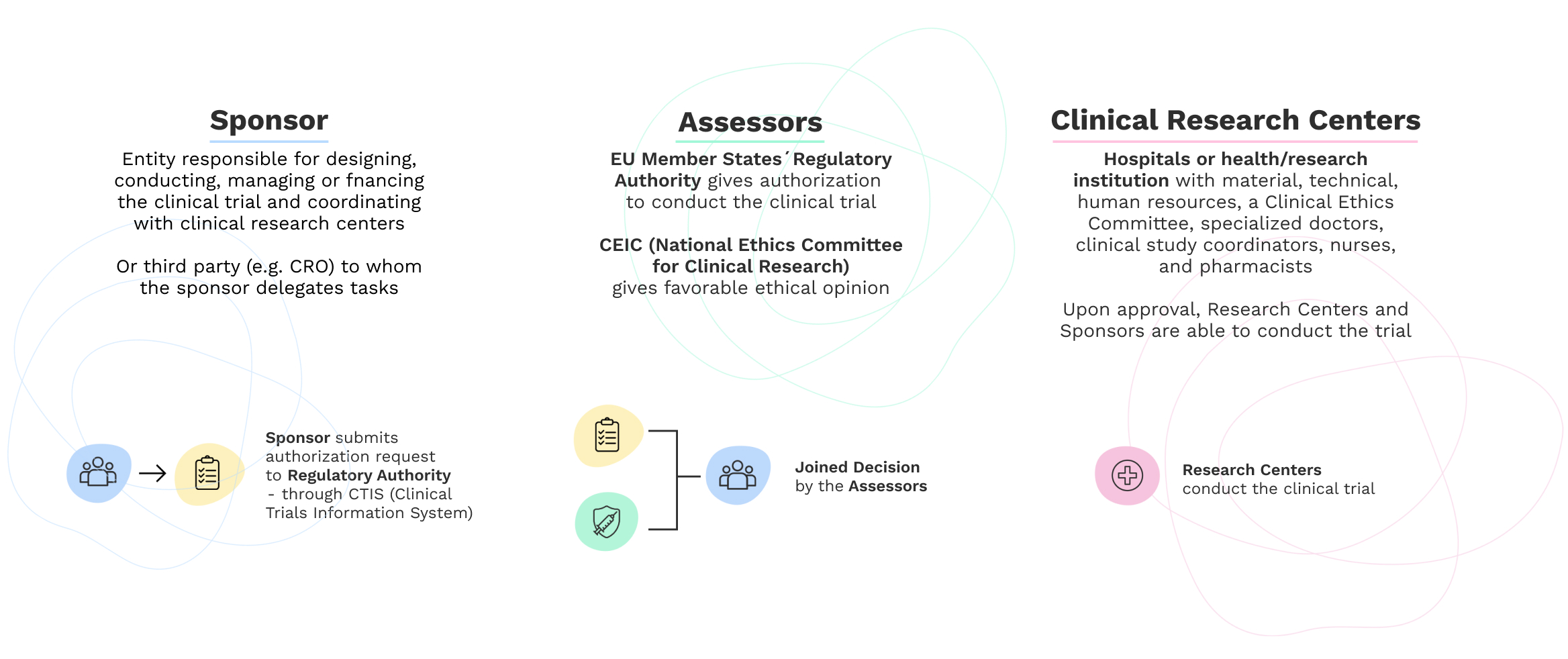
Process of submission and implementation of clinical trials with medicines under the New Regulation 536/2014
The Clinical Trials Regulation, EU Regulation nr 536 came into effect on January 31, 2022.
Among other things, the EU Clinical Trials Regulation changes the system for submitting, assessing and supervising clinical trials in the European Union (EU) and in the countries of the European Economic Area (EEA), that is now carried out through a Portal (EU Portal and Database, EUPD) which is the access point to an integrated European clinical trial information system (Clinical Trials Information System, CTIS) that became operational on the same date.
The CTIS will thus become the single point for submissions, registration of information and issuance of authorizations for clinical trials requests, with restricted access, providing operational support to user processes (both national competent authorities (NCA) and Ethics Committees (EC ) of the Member States (MS) as sponsors).
Table of the transition period:
CTIS also aims to make information about clinical trials in Europe more accessible and transparent. It is thus subject to transparency rules and interacts with 5 European Medicines Agency (EMA) databases, allowing users to search for information on the organizations involved and on the medicines. It also includes an open area for information to citizens and to the general public.
The Regulation revokes the Clinical Trials Directive in a phased approach, by applying a transition period.
Table of the Transition Period
.
Preparation for entry into force of the Regulation
- Consider the transition period in planning the legal framework that is intended to apply or to which the trials will have to move, currently in progress and/or in preparation, taking into account the timing of regulatory activities during the life cycle of the trial.
- Adjust the processes and the management of access/editing/submission rights of CTIS users to the way it is intend to plan and manage the trials in accordance with the Regulation and through CTIS.
- Obtain information and training on the CTIS for the application of the new system, which is supported by extensive programs prepared by the European Medicines Agency (EMA) responsible for its maintenance. These programs are both general in nature and aimed at the different types of entities involved in conducting, assessing, and supervising clinical trials. Thus, it is very important that both commercial and non-commercial/academic sponsors consult the extensive and comprehensive training content that the EMA website makes available.
- Consultation guide of documents published by the EMA, that are relevant for Sponsors and Investigators/academic sponsors, who intend to implement the Clinical Trials Regulation 536/2014 of 16 April in their training processes and programs - Guide Download
- From an operational point of view, consideration must be given to aspects prior to submitting an application to start a clinical trial at CTIS, namely:
- Ensure that the relevant data is registered in the EMA databases and systems with which the CTIS interacts, namely the EMA Account Management, (for users registration), the OMS (Organization Management Service, for registration/research of entities) and the xEVMPD (Extended EudraVigilance medicinal product dictionary, for the registration/research of medicines). The status of the data recorded in the OMS, in case of doubt from the sponsor, can be verified through the SPOR(Substance, product, organization and referential portal) of the EMA.
- Create an EMA account required to access the sponsors restricted work area.
-
- Note: Information on how to register a promoter administrator is available at CTIS online modular training programme, namely modules 3 and 19 and the step-by-step guides and video tutorials (CTIS Highlights newsletter). Registration for CTIS (high-level) administrators has been open since September 2021.
- Ao ser considerado o registo de utilizador no CTIS as organizações devem ponderar se pretendem assumir uma abordagem centrada:
- When performing the user registration in CTIS, organizations should consider whether they intend to take a centered approach
On the organization (organization Centric) or On the Clinical Trial (CT-centric approaches)
Note: Information on how to register a sponsor administrator is available in module 7 of the EMA's CTIS online modular training programme
For more information, refer to the following websites:
Expedited assessment procedure for Clinical Trials with drugs for COVID-19 - 01/02/2022 - 31/01/2024
In the context of the European Union's strategy to respond to the need for investigation of treatments for COVID-19, the European Commission launched a Joint Action (JA), within the scope of EU4Health, for the coordinated and accelerated evaluation of multinational clinical trials for COVID19 as a treatment indication. This evaluation procedure, under the new Clinical Trials Regulation will start from February 2022, with the participation of Portugal.
In order to provide potential sponsors of clinical trials with information about this joint action, an informative session was organized by the JA Coordinator and the Steering Group, aimed at interested parties when they intend to carry out multinational trials.
Video of the session of this action available at:
Clinical investigations of medical devices
For additional information on the submission process of clinical investigations of medical devices see here.
What is a medical device clinical investigation?
Clinical investigation is a systematic investigation involving one or several participants, carried out to assess the safety and performance of a device.
The Medical Devices Regulation 2017/745 (MDR) is applicable from May 26, 2021. This Regulation presents some changes in clinical investigations compared to the previous regulatory framework.
An important change from the RDM is that there are several different types of clinical investigations specifically described in the legislation, including:
- Pre-CE marking clinical investigations: Clinical investigation of medical devices without CE marking (article 62 of the MDR);
- Post-CE-marking clinical investigations: Clinical investigations relating to devices bearing the CE marking or post-marketing follow-up clinical investigations (PMCF) (Article 74 of the MDR);
- Clinical investigations performed to demonstrate compliance of devices, as provided for in article 62 of the MDR (article 82 and additional requirements of national legislation);
- Clinical investigations of medical devices with no intended medical purpose (Annex XVI of the MDR for additional information).
Additional information on the type of clinical investigation and reporting requirements or authorization/opinion requests are available at RNEC. Also relevant is the Q&A document on MDR 2017/745 "MDCG 2021-6 Regulation (EU) 2017/745 - Questions & Answers regarding clinical investigation", available at the European Commission website.
The MDR provides for a centralized European electronic system for submitting clinical investigation requests/notifications (the EUDAMED system). Until this system is operational, submissions in Portugal must be made through the RNEC platform.
For submission of substantial changes to clinical investigations, in accordance with article 75 of the MDR, the applicable section of the RNEC should be consulted.
For the reporting of serious adverse events and medical device defects, as provided for in article 80 of the MDR, the instructions available at the RNEC platform in the applicable section must be followed. Equally applicable to these notifications, the European Commission standard “MDCG 2020 10/1 Safety Reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745)”.
More information on clinical investigations available at:
- ISO 14155 – Clinical Investigations involving medical devices in human subjects
- World Medical Association Declaration of Helsinki on Ethical Principles for Medical Research Involving Human Subjects
- MDCG 2020/10 – Safety Reporting in Clinical Investigations of medical devices under the regulation (EU) 2017/745
- MDCG 2021-6: Regulation (EU) 2017/745 - Questions and Answers regarding clinical investigation